
If I were to ask you to think of a genetic disease that should be a priority for gene therapy research, you might say breast cancer, Huntington’s, or cystic fibrosis. And these are all great candidates. But I’d say you’re missing a big one. Sickle cell disease (SCD) is often overlooked, much like the population it affects, and underfunded, even though it is more common than most household-name diseases.
Did you know that SCD – a genetic disorder of hemoglobin, the oxygen-carrying molecule found in red blood cells (RBCs) – primarily affects those of African descent?
Did you know that SCD is debilitating? The normal rounded disk-like RBCs become “sickled” when cells are deprived of oxygen, and are prone to getting stuck in small blood vessels, causing blockages in blood flow. These blockages (vaso-occlusive events) often result in extreme pain (sickle cell crisis), but can also cause strokes and tissue death in other organs. Patients spend their lives avoiding triggering events like excessive exertion and dehydration. Many require frequent blood transfusions, or repeatedly need treatment for their pain, often causing them to be mislabeled as “drug-seeking.”
Did you know that the mainstay treatment for severe SCD (hydroxyurea) was first used in 1984? But not much has been developed since then (in almost 40 years!). Not to mention it comes with side-effects of bone marrow suppression, affecting white blood cell and platelet production.
Did you know that despite the disparities in awareness and research funding we’re finally seeing some advances and hope?
No?
Well, there has been some very exciting progress in gene therapies that are finishing their first clinical trials. You may have heard of one of them, as it was featured in an interview with SCD researcher Dr. Druv Khullar on CBC’s The Current in March 2022.

To appreciate the brilliance of the genetic engineering approaches, it helps to have a rudimentary understanding of the genetics of SCD. Normal adult hemoglobin is comprised of two a-globin and two b-globin subunits. SCD occurs when a person has inherited two mutated copies (one from each parent) of the b-globin gene, resulting in an abnormal b-globin protein, and ultimately an abnormal hemoglobin molecule: HbS. Under low oxygen conditions the HbS molecule changes from its normal “cloverleaf” shape to a more linear molecule, which causes sickle-shaped RBCs.
bluebirdbio recently published an interim report on a phase I/II clinical trial examining the biological and clinical efficacy of their LentiGlobin infusion. The goal of the LentiGlobin therapy is to engineer a patient’s own haematopoietic stem cells (HSCs) to produce functional hemoglobin. By inserting a gene (human beta-A-T87Q globin) that contains a correction of the sickle cell mutation, cells will produce a hemoglobin molecule with normal morphology that will no longer lead to sickling.
An alternative gene therapy borrows its approach from hydroxyurea, a medication that increases production of fetal hemoglobin (HbF). HbF, produced in utero until about six-weeks after birth, contains g-globin in lieu of b-globin, thus is free of mutations (i.e. no sickling). A recent phase I clinical trial assessed HSCs engineered to produce HbF by blocking the repressor of the g-globin gene, BCL11A. Last year, the group published an interim report on the safety and initial efficacy of modifying a patient’s HSCs to prevent the BCL11A mRNA from being made into a protein (releasing the repression of g-globin).
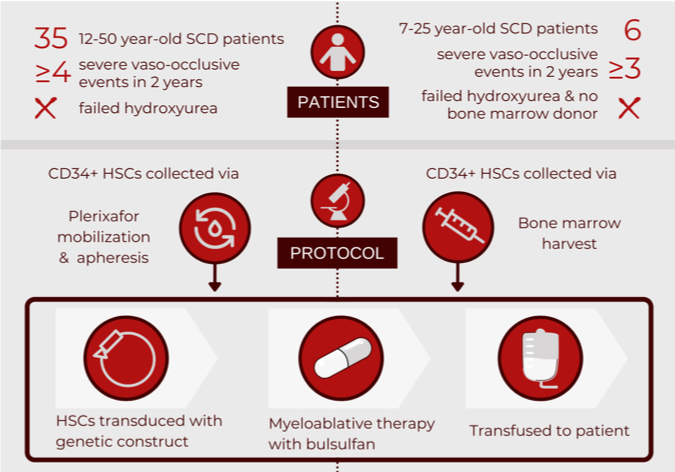
LentiGlobin investigators reported that all patients demonstrated engraftment of the engineered HSCs. Up to three years post-infusion, there was complete resolution of severe vaso-occlusive events in all patients: a clinical cure of SCD. There have been no cases of malignancy related to the LentiGlobin therapy; side effects were limited to the myeloablative chemotherapy. Laboratory outcomes demonstrated a significant increase in patients’ baseline hemoglobin levels, which were largely comprised of the modified hemoglobin. Study participants continue to be followed for cure longevity and any long-term complications.
As for the HbF approach, investigators reported successful engraftment of the engineered HSCs in all patients. Clinical outcomes demonstrated that at 6-18 months post-transfusion, no patients had suffered from severe events such as stroke or vaso-occlusive pain crisis. Side effects were limited to the conditioning chemotherapy. Patients’ baseline hemoglobin levels increased significantly, with HbF comprising almost one third of total levels. Next steps include recruiting more patients for further efficacy testing.

For CRISPR-Cas9 fans: another group of researchers is attempting to increase production of the g-globin gene (and therefore HbF) by using the CRISPR-Cas9 system to edit the enhancing region of the BCL11A gene (essentially turning it “off”). A phase I/II/III clinical trial for this method is just beginning recruitment.
You would think stem cell transplants are an obvious curative option for those with SCD. However, it is quite the opposite. Any donor needs to be a perfect (100%!) match – usually a sibling who has not inherited the disease – which is very rare. In fact, stem cell transplants are so rare, the successful transplant of an adult patient at Toronto’s University Health Network was recently a cause of celebration. Being able to use a patient’s own stem cells would significantly increase the ability to offer chance of a cure. A group from UCLA recently published a comprehensive atlas for human HSC development, which offers the potential to finally make functional blood cells from pluripotent stem cells. This roadmap could highlight additional targets for gene therapy in SCD, and will help overcome the obstacles of requiring bone marrow donors.
So far, these genetic engineering techniques show great promise for the potential cure of SCD. But, as stated in a recent NEJM editorial, we will need to wait for the long-term outcome data to know just how curative these therapies will be. I’m certain that those with SCD will be anxiously awaiting the results, hopeful for the ability to live a normal life free of pain.
However, even a successful curative therapy is not without limitations. Dr. Khullar’s New Yorker article eloquently discusses the challenges with delivering such therapies to marginalized populations in a broken health-care system. (While an American perspective, I believe it holds true in Canada). And these issues will be only more evident in the African populations who suffer from SCD the most: how will such an advanced and expensive therapy be made accessible? Are critics of gene therapy right: will it only serve those few who are lucky enough to access the treatment, rather than the many who cannot?
Sara M. Nolte
Latest posts by Sara M. Nolte (see all)
- Ottawa researchers make tumour-marker negative cancers positive with oncolytic viruses - December 19, 2024
- Cell-based therapy approved as alternative to standard UCB transplant for hematological malignancies - June 21, 2023
- Finding the perfect match: breaking down the science of organ and stem cell transplant matching - April 18, 2023




Comments