
Lyla El-Fayomi is an MSc student in the Molecular Genetics department at the University of Toronto. Under the supervision of Dr. Derek van der Kooy at the Donnelly Centre for Cellular and Biomolecular Research, she is currently studying neural stem cells using single-cell RNA sequencing. She also studies the neurobiology of opiate addiction using optogenetics. For more, follow Lyla on twitter at @LylaElfay.
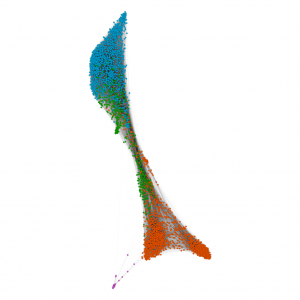
scRNAseq of common myeloid progenitors (green; middle) diverging into populations of erythrocytes (blue; top) and granulocytes (red, orange, pink; bottom). Further sub-clusters can be observed within the orange granulocyte population, including monocytes, neutrophils and basophils. Data were processed in R using Seurat, and then the Scran package was used to construct a kNN graph. At this stage, the file was exported into Gephi, where the ForceAtlas2 layout was used for the final visualization. Public data obtained from Paul et al., 2015. Plot created by David Cook (@DavidPCook).
You may have noticed a dramatic increase in the use of RNA sequencing (RNAseq) over the past few years. Of its many applications, two of the most recognizable include the interrogation of differential gene expression levels and the study of alternative splicing events. In the end, the data always describe homogenized samples as a whole; the need to characterize particular cell types within a sample necessitated the development of a new method to capture information at single-cell resolution.
This need was met by single-cell RNA sequencing (scRNAseq) when it was first published in 2009. It has since been employed in research topics such as stem cell differentiation throughout development, cellular heterogeneity within complex tissues (including tumors!), and the identification of genetic markers for particular cell types.
scRNAseq is an especially important innovation in stem cell research, as stem cells are often mixed in with other cell types in a tissue, and can be relatively rare or difficult to purify.
At the intersection of stem cell research, developmental biology and scRNAseq lies a paper published by Toronto researchers investigating cortical development in mice. First of all, they found that the differentiation of neurons during development is better described as a transcriptional continuum, rather than a step-wise process. Next, they provide an emergence timeline for radial precursor cells (RPCs), which give rise to neural stem cells (NSCs) during development, and define a “core transcriptional identity” that is maintained by RPCs throughout neurogenesis. Finally, they conclude that RPCs seem to drop into a state of quiescence (inactivity) towards the end of neurogenesis, and their aforementioned “core transcriptional identity” appears to be shared with a type of quiescent adult neural stem cell found in the forebrain. This knowledge, made possible by scRNAseq, informs current attempts to understand brain development and to harness the power of NSCs for the purposes of brain injury repair.
But looking at one type of tissue isn’t all that this versatile molecular technique can do.
Back in April, another project aimed to characterize changes in gene expression that cells undergo as they differentiate, in addition to identifying cell types important in regeneration. The researchers opted to study planarians, which are worms that can regenerate their entire body after literally being sliced to pieces (as long as that piece isn’t the very tip of their heads, or their pharynx… fairly decent odds). Using scRNAseq, they created a cell type atlas, in addition to a lineage tree, of every tissue in the organism. Notably, they used RNA velocity – a package for R software that compares the balance of unspliced RNA to spliced RNA – to help them build their lineage and inform the directionality of differentiation. The biological basis is that as a cell starts to differentiate, expression of a certain gene set begins. These transcripts are not spliced immediately, and are characteristic of whichever cell type is up next. Therefore, we’re given a hint of what’s to come in terms of cell fate.
That being said, high-throughput experimental methods such as scRNAseq generate enormous datasets that must be analyzed using seemingly complex code (such as RNA velocity). Today more than ever, advancements in stem cell research are driving bench scientists towards computational biology.
Recognizing that not all stem cell researchers are equipped to take on the computational challenge that is RNAseq, Dr. William Stanford and Gareth Palidwor at the Ottawa Hospital Research Institute partnered with the Stem Cell Network (SCN) to organize a workshop on the analysis of both bulk and single-cell datasets. As a graduate student just entering the field this year, I was given the opportunity to attend.
One of my most important take-aways: With great power comes great responsibility.
Sponsored by SCN and the Ontario Institute for Regenerative Medicine, a major benefit of their curriculum was that it aimed to teach from the ground-up; rather than simply focusing on the analysis, the instructors began with foundational knowledge. Perhaps the most interesting aspect of this workshop was the emphasis placed on how RNAseq can go awry, and making sure that one really understands the tools that they are using. It is a simple concept that should be applied to every field in science, and most might dismiss this as “common sense,” – yet it is still enough of an issue that it needed to be mentioned. Every part of the pipeline, from experimental design to analysis of the data using popular R packages, offers the possibility of misstep (click here for some pretty stellar examples).
Whether you learn the ins and outs yourself or find a collaborator, keeping up with the rapid improvements being made to sequencing platforms and analytical tools currently in existence can be a challenge. That being said, I recently came across a few helpful resources designed to assist in choosing which tools are best for a given study. For instance, this paper compares scRNAseq methods, and this one compares computational methods to infer single-cell trajectories.
Despite the challenges, selecting the right technology and analyzing the data carefully can make scRNAseq an invaluable tool to probe cellular processes at unprecedented resolution. Stem cell biologists across the world are finding ways to add it to their toolkits, and I am certainly looking forward to watching it evolve.
Guest
Latest posts by Guest (see all)
- Regenerative immunotherapy: Hope for chronic autoimmune diseases - September 16, 2025
- Canada’s regenerative revolution: Why AI is the catalyst - September 4, 2025
- Summer by Design: A launchpad for future entrepreneurs and industry scientists - August 14, 2025




Comments